- 移动端


复百澳(苏州)生物医药科技有限公司
8 年
手机商铺
商家活跃:
产品热度:
- NaN
- 0.7999999999999998
- 1.7999999999999998
- 0.7999999999999998
- 3.8
Crispr/cas9 慢病毒包装(承诺)
¥6000 - 10000
推荐产品





公司新闻/正文
实战:不一样的 CRISPR/CAS9 靶点活性验证
5693 人阅读发布时间:2018-06-21 09:29
继慢病毒、溶瘤病毒两个系列专题后,从本期开始以「CRISPR/CAS9 基因编辑技术」为系列专题,分别从 CRISPR/CAS9 靶点活性检测方法、CRISPR/CAS9-sgRNA 文库的设计和运用以及 CRISPR/CAS9 基因编辑技术研究进展三个方面展开。

摘 要
「CRISPR/CAS9 基因编辑技术,已经作为一个常态化的实验技术运用于我们的平常实验中了,其中靶点的有效性验证至关重要,最为经典的检测方法则是 T7E1 酶切鉴定法,该方法对于切割效率较高的靶点,检测效果明显,而对敲除效率不高的靶点,则效果不佳。那么,针对敲除效率不高但又必须要敲除的靶基因,该如何进行敲除效率检测呢?本文介绍了一种基于基因组 PCR 产物的 sanger 法来判定靶点的敲除效率,其灵敏度远高于 T7E1 酶切法!」
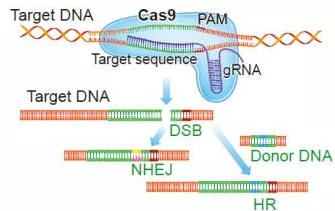
CRISPR(Clustered regularly interspaced short palindromic repeats)规律成簇间隔短回文重复,是源于细菌的一种适应性免疫防御,其作用是清除浸入菌体内的病毒或非本体的 DNA。后来经过科学工作者的研究、改造和优化,可以实现对原核细胞和真核细胞的基因组进行基因的敲除、敲入、激活或者抑制等操作的一项新的生物技术。
CRISPR 的基本结构由三个部分组成,即,一个前导区 (Leader)、多个短而高度保守的重复序列区 (Repeat) 和多个间隔区 (Spacer) 组成。(1)前导区 (Leader),位于 CRISPR 结构的上游区域,序列长度在 300~500bp 之间,其中 AT 含量较高是启动子序列;(2)重复序列区 (Repeat),序列长度为 21~48bp,含有回文序列,可形成发卡结构;(3)间隔区 (Spacer),该序列区穿插在重复序列区 (Repeat) 之间,序列长度在 26~72bp 的之间。
CRISPR 工作原理是 CRISPR-derived RNA (crRNA) 通过碱基配对与 tracrRNA (trans-activating RNA) 结合形成 tracrRNA/ crRNA 复合体,此复合体招募 CRISPR 核酸酶聚集于 crRNA 配对的序列靶位点处,并且对靶位点 DNA 双链进行切割,细胞通过同源修复机制对断裂后的 DNA 双链进行修复,研究者可根据同源修复机制的原理,设计相关的实验对靶基因进行编辑(如:去除和/或添加新基因)。
02
T7E1 酶切鉴定法
T7 核酸内切酶 I (T7E1)识别并切割不完全配对 DNA、十字型结构 DNA、Holliday 结构或交叉 DNA、异源双链 DNA 或者以更慢的速度切割或切刻双链 DNA。该酶切割错配碱基 5′ 端的第一、第二或第三个磷酸二酯键。
在 CRISPR/CAS9 阳性克隆检测中常会出现——假阳性和弱阳性。
假阳性多由于 T7E1 酶识别了非 CRISPR/CAS9 切割修复导致的碱基错配,譬如,识别了十字交叉,holiday 结构等。
弱阳性,主要是由于靶点 gDNA 的序列结构复杂或异常导致的。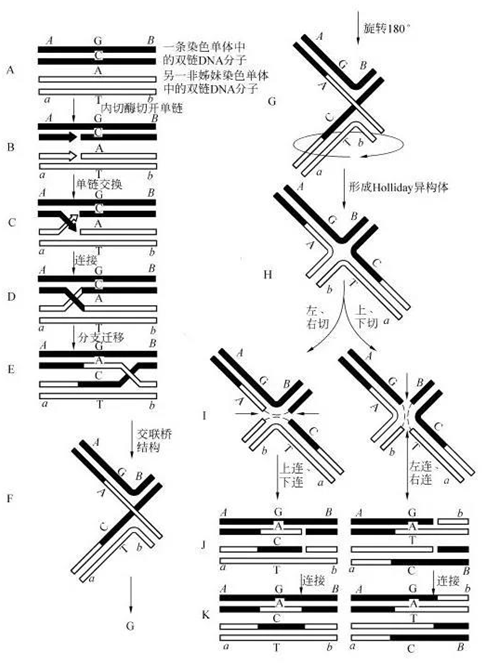
● T7E1 酶切法检测突变体实验步骤
I. PCR 扩增出带有突变位点 (如 TALEN or CRISPR/Cas9 的 target site)DNA 片段,长度约 500bp,突变位点最好不位于 PCR 片段中央,这样将切割出两条大小不同的带。
II. 以混合克隆细胞株 gDNA(或者突变体 DNA 与野生型 DNA 混合)为模板,经过 PCR 扩增后的产物,按如下体系进行混合,进行加热变性、退火复性处理。
III. 上述产物用 T7E1 酶的酶切处理后(37℃ 水浴, 1 h~1.5 h),用 2% agrose DNA 电泳鉴定检测。如可以切出两条带的为有效靶点,反之为无效靶点,有效靶点电泳如下图。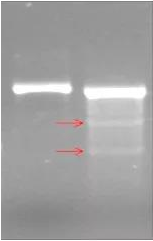
Sanger 法测序是根据核苷酸在某一固定的点开始,随机在某一个特定的碱基处终止,并且在每个碱基后面进行荧光标记,产生以 A、T、C、G 结束的四组不同长度的一系列核苷酸,然后在尿素变性的 PAGE 胶上电泳进行检测,从而获得可见 DNA 碱基序列的一种方法。在分子生物学研究中,DNA 的序列分析是进一步研究和改造目的基因的基础。目前用于 DNA 测序的技术主要有 Frederick Sanger 发明的 Sanger 双脱氧链终止法(Chain Termination Method)。
04
实战:不一样的 CRISPR/CAS9 靶点活性验证
针对目的基因或序列的 sgRNA 构建完毕后,都需要进行该位点的活性检测实验,对于高敲除效率的靶点当然不用担心后续实验问题(如下图泳道 2),但对于靶点敲除效率不高的咋办呢?(如下图泳道 4)
对于假阳性或弱阳性是否有其他的检测方法?
亦或是否有其他简洁、经济、有效的敲除效率检测方法?
有!PCR 产物 Sanger 测序!
1. A 基因 CON;2. A 基因 T7E1 酶切;3.B 基因 CON;4. B 基因 T7E1 酶切 ; 5. 实验阳性对照。
● Sanger 测序验证敲除有效流程
gDNA 模板 PCR 产物测序验证法和用 T7E1 酶切的验证方法的前期实验和要求路线基本一致,PCR 扩增出带有突变位点 (如 TALEN or CRISPR/Cas9 的 target site)DNA 片段,长度约 500bp,突变位点最好不位于 PCR 片段中央,这样将切割出两条大小不同的带。PCR 结束后无需进行退火等步骤直接用 PCR 扩增引物进行 Sanger 测序验证。
● PCR 产物测序,发现你所没有发现
PCR 产物测序,测序引物为 PCR 扩增时的正向或反向引物。
测序结果如下:
上行为 gDNA 序列,其中大写字母部分为靶点,下行为测序结果。这个结果比对,着实伤心!什么突变啊,什么敲除啊,啥子都没有…………
咋办?
不急再看看下面这个图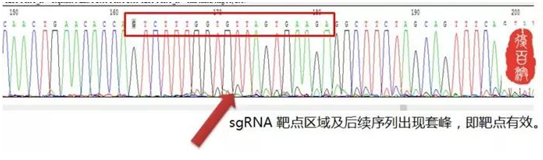
红色框部分为靶点序列,在靶点序列后面出现了杂峰,套峰!
这说明什么?说明这个靶点是有效的,此处,产生了切割、敲除、NHEJ……
喔喔喔……,看到了希望的曙光!
但是,这么低效率如何才能筛选出基因敲除的细胞呢?
● 病毒感染,绝处逢生
导致敲除效率不高的原因除了设计的靶点效率不高外,还有就是 sgRNA-CAS9 转导进入细胞的效率较低。将 sgRNA-CAS9 包装进入病毒载体,通过病毒感染目的细胞,提高 sgRNA&CAS9 的表达量,可以有效的提高 CAS9 蛋白对目的基因的切割效率。如下图:
1.B 基因 CON;2. B 基因 T7E1 酶切
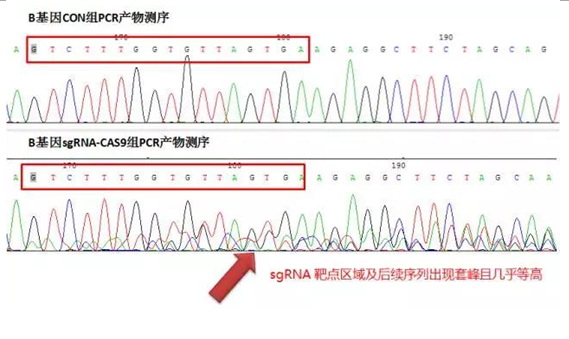
由结果分析可以看出,用 sgRNA-CAS9 病毒载体感染细胞后,即使用 T7E1 酶切检测的方法也可以明显的检测出基因的敲除效率。用 gDNA 模板 PCR 产物测序检测方法,也可以通过峰图中套峰的高低来判定 sgRNA-CAS9 的敲除效率。
通过合理的实验流程设计,可以很好节约实验时间,提高实验效率,百澳笑笑生的实验流程如下:
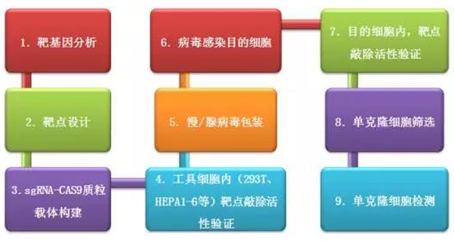
说明:
第 4 步,工具细胞内(293T、HEPA1-6 等)靶点敲除活性验证,本步骤旨在根据需要进行敲除的细胞属性(human、mouse 等)选择合适的、转染效率较高的工具细胞,进行靶点敲除效率验证实验。
第 7 步,目的细胞内,靶点敲除活性验证,本步骤旨在复核验证靶点敲除的有效性,并对后期的单克隆细胞的筛选库的数量做出预判,如果敲除效率较高,在后期的单克隆筛选时可以适当减少筛选细胞数量,反之,则需要增加筛选细胞的数量。
第 9 步,单克隆细胞检测,本步骤旨在检测基因敲除的结果,看是否将目的基因 KO,检测不出其表达,通常用 WB 检测。此处建议增加一个 gDNA 测序检测,确认获得的 KO 细胞是纯合子还是杂合子。如下图:
B 基因 KO 纯合子(单峰)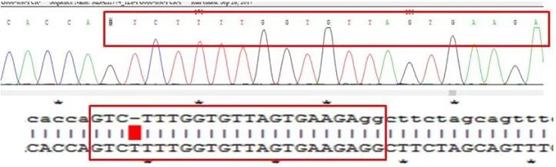
B 基因 KO 杂合子(套峰)
06
附: 单克隆细胞筛选实验步骤
实验步骤
1. 将目的细胞接种于 6 孔板中至 70-80% 密度 ;
2. 待细胞贴壁后,加入 puromycin 药物进行空细胞致死最低浓度筛选,一般药物浓度梯度设置为 1ug/ml、2.5ug/ml、5ug/ml、10ug/ml(终浓度)进行筛选;
3. 药物处理 48 h 后细胞全部死亡的最低药物浓度即后续稳定株筛选的药物使用终浓度 ;
4. 将目的细胞接种于 6 孔板中,待细胞贴壁后进行慢病毒感染,感染后第二天,加入最低浓度的 puromycin 药物进行筛选,同时做一份空细胞作为对照。显微镜下观察存活细胞中示踪基因的表达(puromycin 或 GFP), 继续维持培养;
5. 待细胞密度生长至 90% 时,常规消化后接种于 96 孔板中,调整接种的细胞密度为 1-1.5 个/孔(或者用流式细胞仪直接进行单克隆细胞分选);
6. 继续培养两周左右,期间适时补液或换液,待孔板中细胞长至细胞克隆时,挑取阳性细胞克隆至 24 孔板中扩大培养并检测目的敲除(PCR 测序或 WB 检测);
7. 正确克隆进一步的扩大培养,直至获得需要的细胞量。
参考资料
1. Genome-scale CRISPR-Cas9 knockout screening in human cells. Shalem O*, Sanjana NE*, Hartenian E, ShiX, Scott DA, Mikkelsen T, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F (2014). Science, 343, 83-7. DOI:10.1126/science.1247005
摘 要
「CRISPR/CAS9 基因编辑技术,已经作为一个常态化的实验技术运用于我们的平常实验中了,其中靶点的有效性验证至关重要,最为经典的检测方法则是 T7E1 酶切鉴定法,该方法对于切割效率较高的靶点,检测效果明显,而对敲除效率不高的靶点,则效果不佳。那么,针对敲除效率不高但又必须要敲除的靶基因,该如何进行敲除效率检测呢?本文介绍了一种基于基因组 PCR 产物的 sanger 法来判定靶点的敲除效率,其灵敏度远高于 T7E1 酶切法!」
01
CRISPR/CAS9 技术简介
CRISPR/CAS9 技术简介
CRISPR(Clustered regularly interspaced short palindromic repeats)规律成簇间隔短回文重复,是源于细菌的一种适应性免疫防御,其作用是清除浸入菌体内的病毒或非本体的 DNA。后来经过科学工作者的研究、改造和优化,可以实现对原核细胞和真核细胞的基因组进行基因的敲除、敲入、激活或者抑制等操作的一项新的生物技术。
CRISPR 的基本结构由三个部分组成,即,一个前导区 (Leader)、多个短而高度保守的重复序列区 (Repeat) 和多个间隔区 (Spacer) 组成。(1)前导区 (Leader),位于 CRISPR 结构的上游区域,序列长度在 300~500bp 之间,其中 AT 含量较高是启动子序列;(2)重复序列区 (Repeat),序列长度为 21~48bp,含有回文序列,可形成发卡结构;(3)间隔区 (Spacer),该序列区穿插在重复序列区 (Repeat) 之间,序列长度在 26~72bp 的之间。
CRISPR 工作原理是 CRISPR-derived RNA (crRNA) 通过碱基配对与 tracrRNA (trans-activating RNA) 结合形成 tracrRNA/ crRNA 复合体,此复合体招募 CRISPR 核酸酶聚集于 crRNA 配对的序列靶位点处,并且对靶位点 DNA 双链进行切割,细胞通过同源修复机制对断裂后的 DNA 双链进行修复,研究者可根据同源修复机制的原理,设计相关的实验对靶基因进行编辑(如:去除和/或添加新基因)。
02
T7E1 酶切鉴定法
T7 核酸内切酶 I (T7E1)识别并切割不完全配对 DNA、十字型结构 DNA、Holliday 结构或交叉 DNA、异源双链 DNA 或者以更慢的速度切割或切刻双链 DNA。该酶切割错配碱基 5′ 端的第一、第二或第三个磷酸二酯键。
在 CRISPR/CAS9 阳性克隆检测中常会出现——假阳性和弱阳性。
假阳性多由于 T7E1 酶识别了非 CRISPR/CAS9 切割修复导致的碱基错配,譬如,识别了十字交叉,holiday 结构等。
弱阳性,主要是由于靶点 gDNA 的序列结构复杂或异常导致的。
● T7E1 酶切法检测突变体实验步骤
I. PCR 扩增出带有突变位点 (如 TALEN or CRISPR/Cas9 的 target site)DNA 片段,长度约 500bp,突变位点最好不位于 PCR 片段中央,这样将切割出两条大小不同的带。
II. 以混合克隆细胞株 gDNA(或者突变体 DNA 与野生型 DNA 混合)为模板,经过 PCR 扩增后的产物,按如下体系进行混合,进行加热变性、退火复性处理。
III. 上述产物用 T7E1 酶的酶切处理后(37℃ 水浴, 1 h~1.5 h),用 2% agrose DNA 电泳鉴定检测。如可以切出两条带的为有效靶点,反之为无效靶点,有效靶点电泳如下图。
03
Sanger 测序
Sanger 测序
Sanger 法测序是根据核苷酸在某一固定的点开始,随机在某一个特定的碱基处终止,并且在每个碱基后面进行荧光标记,产生以 A、T、C、G 结束的四组不同长度的一系列核苷酸,然后在尿素变性的 PAGE 胶上电泳进行检测,从而获得可见 DNA 碱基序列的一种方法。在分子生物学研究中,DNA 的序列分析是进一步研究和改造目的基因的基础。目前用于 DNA 测序的技术主要有 Frederick Sanger 发明的 Sanger 双脱氧链终止法(Chain Termination Method)。
04
实战:不一样的 CRISPR/CAS9 靶点活性验证
针对目的基因或序列的 sgRNA 构建完毕后,都需要进行该位点的活性检测实验,对于高敲除效率的靶点当然不用担心后续实验问题(如下图泳道 2),但对于靶点敲除效率不高的咋办呢?(如下图泳道 4)
对于假阳性或弱阳性是否有其他的检测方法?
亦或是否有其他简洁、经济、有效的敲除效率检测方法?
有!PCR 产物 Sanger 测序!
1. A 基因 CON;2. A 基因 T7E1 酶切;3.B 基因 CON;4. B 基因 T7E1 酶切 ; 5. 实验阳性对照。
● Sanger 测序验证敲除有效流程
gDNA 模板 PCR 产物测序验证法和用 T7E1 酶切的验证方法的前期实验和要求路线基本一致,PCR 扩增出带有突变位点 (如 TALEN or CRISPR/Cas9 的 target site)DNA 片段,长度约 500bp,突变位点最好不位于 PCR 片段中央,这样将切割出两条大小不同的带。PCR 结束后无需进行退火等步骤直接用 PCR 扩增引物进行 Sanger 测序验证。
● PCR 产物测序,发现你所没有发现
PCR 产物测序,测序引物为 PCR 扩增时的正向或反向引物。
测序结果如下:
上行为 gDNA 序列,其中大写字母部分为靶点,下行为测序结果。这个结果比对,着实伤心!什么突变啊,什么敲除啊,啥子都没有…………
咋办?
不急再看看下面这个图
红色框部分为靶点序列,在靶点序列后面出现了杂峰,套峰!
这说明什么?说明这个靶点是有效的,此处,产生了切割、敲除、NHEJ……
喔喔喔……,看到了希望的曙光!
但是,这么低效率如何才能筛选出基因敲除的细胞呢?
● 病毒感染,绝处逢生
导致敲除效率不高的原因除了设计的靶点效率不高外,还有就是 sgRNA-CAS9 转导进入细胞的效率较低。将 sgRNA-CAS9 包装进入病毒载体,通过病毒感染目的细胞,提高 sgRNA&CAS9 的表达量,可以有效的提高 CAS9 蛋白对目的基因的切割效率。如下图:
1.B 基因 CON;2. B 基因 T7E1 酶切
由结果分析可以看出,用 sgRNA-CAS9 病毒载体感染细胞后,即使用 T7E1 酶切检测的方法也可以明显的检测出基因的敲除效率。用 gDNA 模板 PCR 产物测序检测方法,也可以通过峰图中套峰的高低来判定 sgRNA-CAS9 的敲除效率。
05
补充:系统设计实验流程,助力实验效率提升
补充:系统设计实验流程,助力实验效率提升
通过合理的实验流程设计,可以很好节约实验时间,提高实验效率,百澳笑笑生的实验流程如下:
说明:
第 4 步,工具细胞内(293T、HEPA1-6 等)靶点敲除活性验证,本步骤旨在根据需要进行敲除的细胞属性(human、mouse 等)选择合适的、转染效率较高的工具细胞,进行靶点敲除效率验证实验。
第 7 步,目的细胞内,靶点敲除活性验证,本步骤旨在复核验证靶点敲除的有效性,并对后期的单克隆细胞的筛选库的数量做出预判,如果敲除效率较高,在后期的单克隆筛选时可以适当减少筛选细胞数量,反之,则需要增加筛选细胞的数量。
第 9 步,单克隆细胞检测,本步骤旨在检测基因敲除的结果,看是否将目的基因 KO,检测不出其表达,通常用 WB 检测。此处建议增加一个 gDNA 测序检测,确认获得的 KO 细胞是纯合子还是杂合子。如下图:
B 基因 KO 纯合子(单峰)
B 基因 KO 杂合子(套峰)
06
附: 单克隆细胞筛选实验步骤
实验步骤
1. 将目的细胞接种于 6 孔板中至 70-80% 密度 ;
2. 待细胞贴壁后,加入 puromycin 药物进行空细胞致死最低浓度筛选,一般药物浓度梯度设置为 1ug/ml、2.5ug/ml、5ug/ml、10ug/ml(终浓度)进行筛选;
3. 药物处理 48 h 后细胞全部死亡的最低药物浓度即后续稳定株筛选的药物使用终浓度 ;
4. 将目的细胞接种于 6 孔板中,待细胞贴壁后进行慢病毒感染,感染后第二天,加入最低浓度的 puromycin 药物进行筛选,同时做一份空细胞作为对照。显微镜下观察存活细胞中示踪基因的表达(puromycin 或 GFP), 继续维持培养;
5. 待细胞密度生长至 90% 时,常规消化后接种于 96 孔板中,调整接种的细胞密度为 1-1.5 个/孔(或者用流式细胞仪直接进行单克隆细胞分选);
6. 继续培养两周左右,期间适时补液或换液,待孔板中细胞长至细胞克隆时,挑取阳性细胞克隆至 24 孔板中扩大培养并检测目的敲除(PCR 测序或 WB 检测);
7. 正确克隆进一步的扩大培养,直至获得需要的细胞量。
参考资料
1. Genome-scale CRISPR-Cas9 knockout screening in human cells. Shalem O*, Sanjana NE*, Hartenian E, ShiX, Scott DA, Mikkelsen T, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F (2014). Science, 343, 83-7. DOI:10.1126/science.1247005